
Adventures in mRNA lipid nanoparticle formulation
Lipid nanoparticles (LNPs) encapsulating neoantigen-encoding mRNA hold promise for the development of more potent therapeutic cancer vaccines. As a lab focused on optimizing personalized cancer treatments, establishing that we are able to formulate and effectively utilize such particles would be a step towards initiating a clinical trial, and the first stages of this translational process often involve generating data that demonstrate efficacy in vitro. This blog post chronicles those endeavors.
An internet search of the term “nanoparticle” will yield diverse results and may lead down a rabbit hole of spherical diagrams with minimal discernible differences, so let’s begin there and climb out together. The specific nanoparticles our lab was interested in formulating contain mRNA along with four essential lipid components: an ionizable lipid, a helper lipid, cholesterol, and a polyethylene glycol (PEG) lipid. Perhaps the most important of these is the ionizable lipid which, as its name suggests, is positively charged at a low pH and neutral at physiological pH. When positively charged, this lipid plays a critical role in interacting with negatively charged mRNA to facilitate particle formation. Additionally, the ionizable lipid promotes entry into the cytoplasm by escaping from the acidic endosome, or the compartment within the cytoplasm where the particle ends up following cellular internalization.
On the flip side of this, the ionizable lipid’s neutral charge at physiological pH is also critical as it makes ionizable lipids less toxic than previously used, permanently charged cationic lipids. Cholesterol and the helper lipid stabilize and support the nanoparticles, while the PEG lipid prevents particles from aggregating. It’s important to note here that all of these components play additional roles in vivo that can affect where these particles go and how long they persist, so this is a rather simplified overview for the sake of brevity. (If you aren’t claustrophobic and want to go further down the rabbit hole, a nice detailed review can be found here.)
Besides the aforementioned lipids, our nanoparticles also contain nucleoside-modified mRNA, which has structural changes to render it less immunogenic. This may sound counterintuitive when the ultimate goal of administering these particles is to stimulate an immune response, but detection of foreign nucleic acids can trigger cellular changes that prematurely halt protein synthesis, preventing the desired outcome.
To formulate the LNPs, we used a benchtop microfluidic device to rapidly mix the lipid and mRNA solutions then dialyzed the mixture overnight, concentrated it, and sterile-filtered for use in cell cultures. Following formulation, we determined the mRNA concentration and encapsulation efficiency along with the particle size and polydispersity index (PDI). All batches produced had an average diameter of between 75 and 150 nm with a PDI of 0.300 or lower and an encapsulation efficiency of at least 95%.
At this point the reader is probably dying to know what these particles would be used for or, if you are like my cat, has fallen asleep. The purpose of the planned experiments was two-fold: demonstrate the ability of antigen presenting cells to take up the mRNA-containing particles and translate the protein, and confirm that antigen presenting cells treated with said particles could stimulate an antigen-specific T cell response. Specifically, we planned to treat dendritic cells, which are potent professional antigen presenting cells, with LNPs containing equal amounts of luciferase- and ovalbumin-encoding mRNA. Protein synthesis would be determined by measuring luminescence following administration of the luciferase substrate, and T cell response would be derived from CFSE dilution of labeled OT-1 cells in co-culture, indicating proliferation upon recognition of their cognate peptide, the ovalbumin epitope SIINFEKL.
Before diving into the results of this set of experiments, I should highlight some of the lessons learned from my first attempts to work with our LNPs in vitro. First, serum concentration matters. In an early experiment working with a cancer cell line, raising the final serum media concentration present in the wells during nanoparticle administration from five to ten percent, which is commonly used in cell culture, reduced luminescence seven-fold, nearly to the level of untreated cells.
Second, in our hands, it was difficult to work with spleen-derived dendritic cells. Besides the yield constraints (only ~0.5-2% of splenocytes are eluted as dendritic cells when using commercially available column-based negative selection kits), I was unable to convincingly transfect these cells. This was regardless of serum concentration, cell number, and the presence or absence of GM-CSF, and it was not due to decreased viability.
Fortunately, bone marrow-derived dendritic cells (BMDCs) provided an alternative antigen presenting cell type that was better suited for these proof-of-concept experiments. In addition to much greater yield per mouse (> 20 million immature BMDCs on the fifth day of culture), BMDCs could be successfully transfected at various time points during differentiation and could be cryopreserved before or after differentiation and then transfected. Additionally, with our specific LNP formulation, serum-free RPMI worked better than PBS or Opti-MEM as diluent for administering treatments 1:1 to BMDCs in serum-containing media.
Finally, I noticed that as particles grew older, there was a corresponding decrease in luminescence. This could not be explained by changes in mRNA concentration or encapsulation efficiency over time or by the formation of large particle aggregates, which were absent when the LNPs were reassessed. However, an easy remedy to this predicament was to use the particles as soon as possible after formulation.
Post-troubleshooting, the experimental design to evaluate protein synthesis was as follows: Murine bone marrow was collected and cultured with GM-CSF in a low-binding six-well plate for five days to differentiate BMDCs. Cells were then plated in a 96-well plate (200,000 cells per well), rested overnight, and treated with LNPs ~12 hours later. Luminescence was measured 24 hours after adding the LNPs. Results, shown in Figure 1, demonstrated that the luciferase protein was present following treatment with any of the LNP doses tested. Though not directly measured, we can be fairly confident that the steps preceding translation also occurred: our LNPs were taken up by the BMDCs, then mRNA was released into the cytoplasm, where it was translated into protein. The next step was to determine whether or not these proteins were processed and presented adequately to stimulate a T cell response.
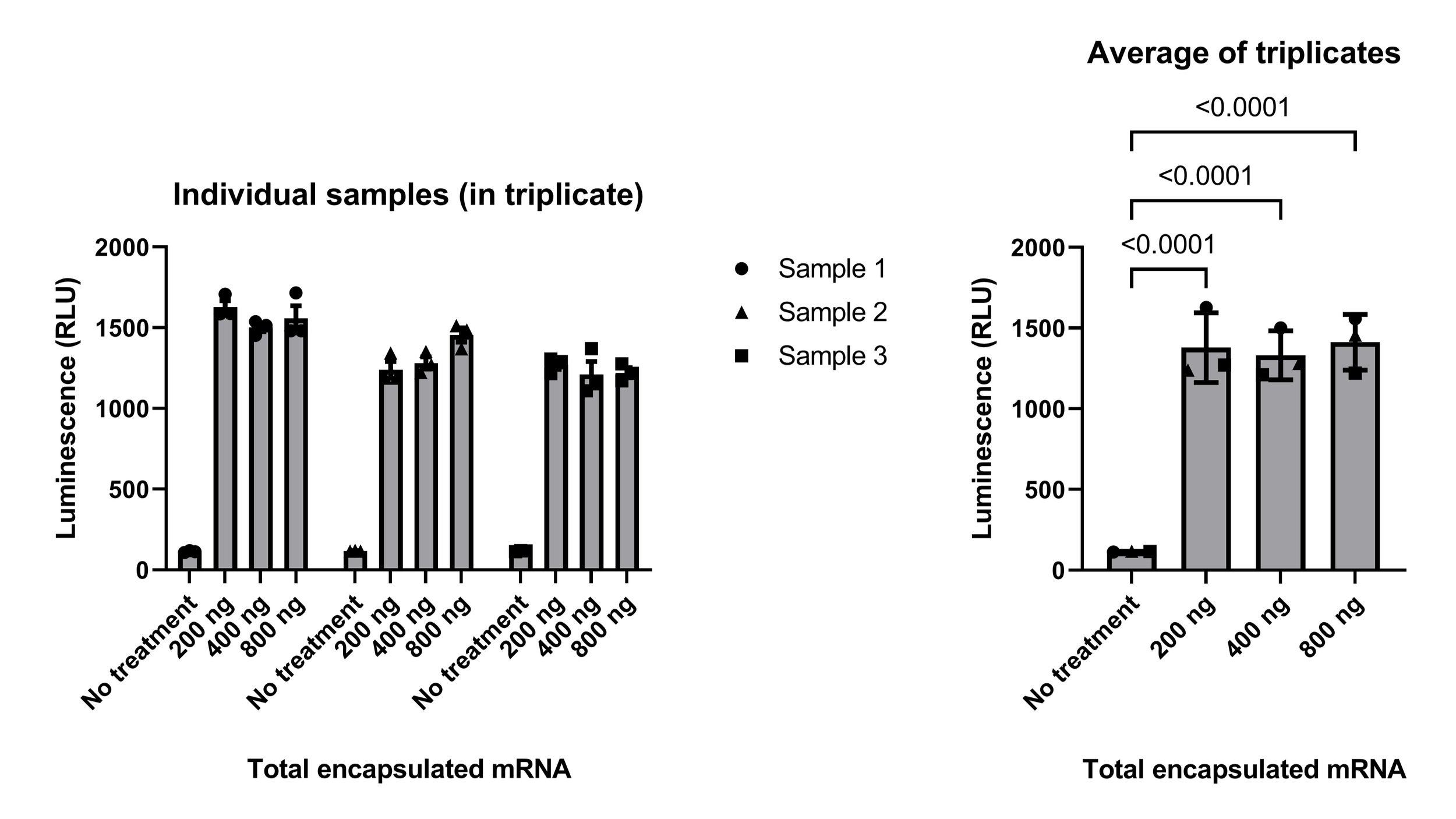
Figure 1 - Results of experiments testing the ability of bone marrow-derived dendritic cells (BMDCs) to synthesize luciferase following treatment with lipid nanoparticles containing luciferase- and ovalbumin-encoding mRNA. Each “sample” is a different BMDC donor mouse (n = 3). Samples were treated in triplicate (left) and triplicates were averaged for analysis (right). Results were analyzed by one-way ANOVA (p < 0.0001) with significant (p < 0.05) Tukey’s multiple comparisons p-values indicated. RLU, relative light unit.
To address this question, I proceeded to the portion of the project where the ovalbumin mRNA gets the spotlight. OT-1 cells, which are transgenic CD8+ T cells bearing receptors that recognize the SIINFEKL peptide (derived from the ovalbumin protein) when bound to the H-2Kb MHC class I molecule, were a useful immunological tool for these experiments. The design was such that OT-1 cells would be isolated from splenocytes, labeled with CFSE, and then cultured with LNP-pulsed BMDCs. Whether the T cells responded to antigen presentation by the BMDCs would be determined using flow cytometry, where a dilution of CFSE indicates proliferation. Similar to those above, these experiments also required some troubleshooting, mainly to determine an adequate number of each cell type (BMDCs and OT-1 cells) to include per well. Initial attempts revealed that 200,000 of each per well was too many and 50,000 BMDCs plus 75,000 OT-1 cells was not enough.
To incorporate these findings, the following experimental setup was used: Cryopreserved bone marrow was thawed and cultured with GM-CSF in a low-binding six-well plate for five days to differentiate BMDCs. These cells were then plated in a 96-well plate (100,000 cells per well), rested overnight, and treated with LNPs ~15 hours later. Six hours after adding the LNPs, cryopreserved isolated CD8+ OT-1 cells were thawed, labeled with CFSE, and added to the culture (110,000-126,000 cells per well). The co-culture incubated for three days, with half of the media replenished on the second day, and was then stained and acquired on a flow cytometer. Figure 2 shows the flow cytometry gating strategy of a representative sample, and the results of two experiments (each using T cells from a different mouse) are below in Figure 3. These results demonstrate a dose-response relationship between mRNA LNP dose and T cell proliferation, wherein higher concentrations of mRNA led to greater proliferation, indicating that ovalbumin is not only being translated within the antigen presenting cells but is also being processed internally and presented to T cells on major histocompatibility complex molecules.

Figure 2 - Gating of flow cytometry samples. Alive and CD3e+ T cell gates were drawn based on fluorescence-minus-one controls.

Figure 3 - Results of experiments testing the ability of bone marrow-derived dendritic cells (BMDCs) to stimulate proliferation of OT-1 T cells following treatment with lipid nanoparticles (LNPs) containing luciferase- and ovalbumin-encoding mRNA. Each panel shows the T cell population defined as in Figure 2 following culture with BMDCs from a different donor mouse (n = 3) treated as described (left).
This brings us to the end of this set of experiments, although work on this project is far from over. Stay tuned to the PIRL blog for future updates, which we anticipate will involve testing the efficacy of mRNA LNP vaccines in vivo.