
Recent Publications

MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing
Tim O’Donnell, Alex Rubinsteyn, Uri Laserson
This paper presents an extensions of our peptide-MHC binding predictor MHCflurry to explicitly consider allele-invariant sequence motifs that correspond to signatures of antigen processing by the proteasome, TAP, and ERAP. The antigen processing score is combined with an allele specific MHC affinity score to derive a combined presentation score.

Alternative tumour-specific antigens
Christof C. Smith, Sara R. Selitsky, Shengjie Chai, Paul M. Armistead, Benjamin G. Vincent, Jonathan S. Serody
The most commonly studied class of tumor specific antigens are those derived from non-synonymous single-nucleotide variants (SNVs), or SNV neoantigens. To increase the repertoire of available therapeutic targets, antigens derived from mutational frameshifts, splice variants, gene fusions, endogenous retroelements and other processes are appealing. In this Opinion article, we outline the biology, available computational tools, preclinical and/or clinical studies and relevant cancers for each alternative tumor specific antigen class. We also discuss both current challenges preventing the therapeutic application of alternative tumor specific antigens and potential solutions to aid in their clinical translation.
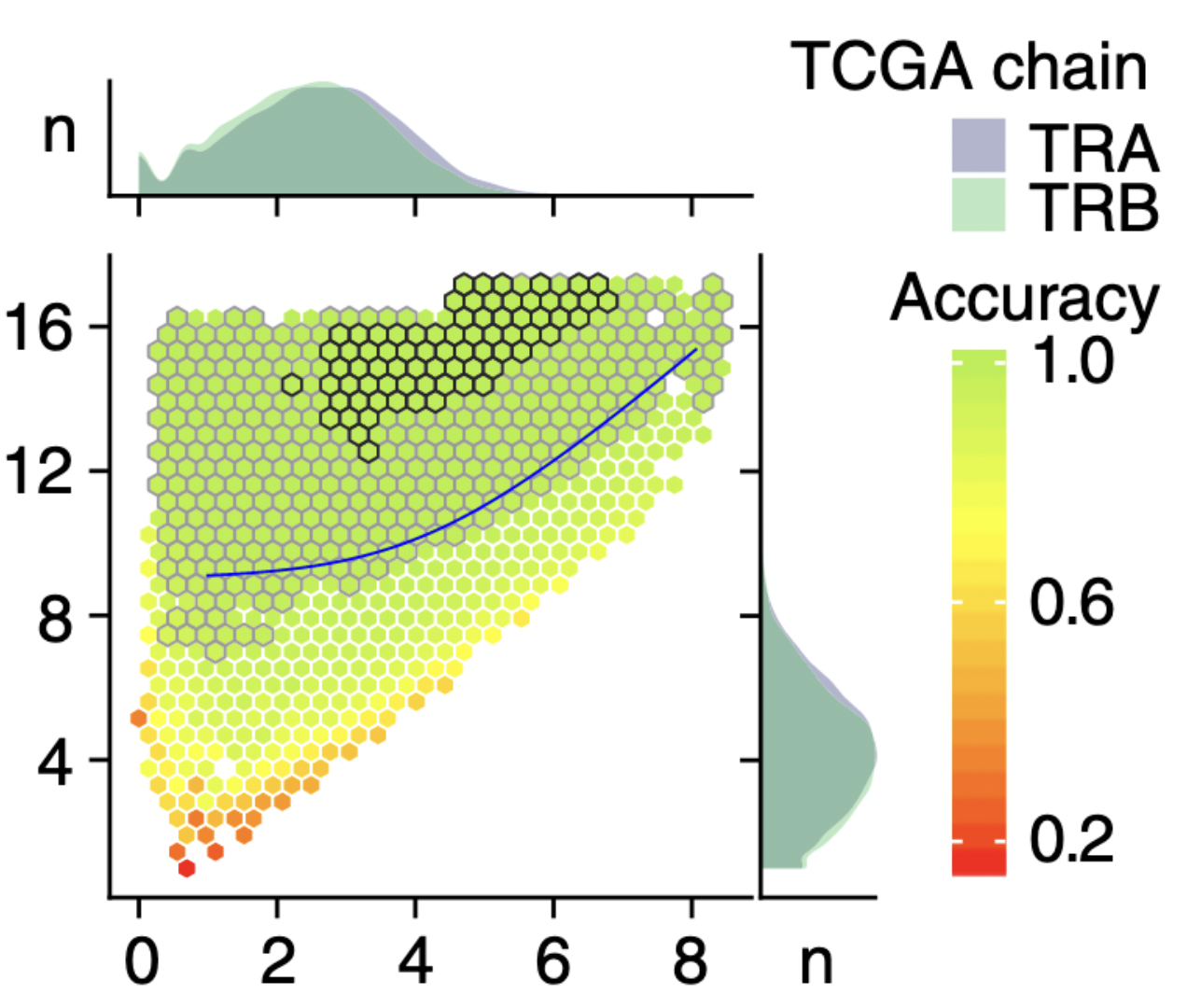
Improved T-cell Receptor Diversity Estimates Associate with Survival and Response to Anti–PD-1 Therapy
Dante S. Bortone, Mark G. Woodcock, Joel S. Parker and Benjamin G. Vincent
We examine the performance characteristics of methods for T-cell receptor repertoire profiling from RNA-sequencing data and found under-sampling as a prominent source of bias in diversity estimates. We then derived a model via statistical learning that attenuates bias to produce corrected diversity estimates. This modeled diversity improved discrimination in The Cancer Genome Atlas data and associated with survival and treatment response in patients with melanoma treated with anti–PD-1 therapy, where the commonly used diversity normalizations did not. These findings have the potential to increase our understanding of the tumor immune microenvironment and improve the accuracy of predictions of patient responses to immunotherapy.

Landscape and Selection of Vaccine Epitopes in SARS-CoV-2
Christof C. Smith, Sarah Entwistle, Caryn Willis, Steven Vensko, Wolfgang Beck, Jason Garness, Maria Sambade, Eric Routh, Kelly Olsen, Julia Kodysh, Timothy O’Donnell, Carsten Haber, Kirsten Heiss, Volker Stadler, Erik Garrison, Oliver C. Grant, Robert J. Woods, Mark Heise, Benjamin G. Vincent, Alex Rubinsteyn
We combine computational prediction of T cell epitopes, recently published B cell epitope mapping studies, and epitope accessibility to select candidate peptide vaccines for SARS-CoV-2. In addition to predicted MHC affinity, candidate T cell epitopes were refined by predicted immunogenicity, viral source protein abundance, sequence conservation, coverage of high frequency HLA alleles and co-localization of CD4+ and CD8+ T cell epitopes. B cell epitope regions were chosen from linear epitope mapping studies of convalescent patient serum, followed by filtering to select regions with surface accessibility, high sequence conservation, spatial localization near functional domains of the spike glycoprotein, and avoidance of glycosylation sites. By combining B cell and T cell analyses, as well as a manufacturability heuristic, we propose a set of SARS-CoV-2 vaccine peptides for use in subsequent murine studies and clinical trials.