
Shaking the Rust Off Python
TL;DR
Dynamic scripting languages such as Python and R are convenient for implementing bioinformatics algorithms. However, these languages incur a significant overhead on every operation, which makes processing read-level data in large quantities slow. One way to speed up a slow algorithm is to move the bottleneck out of Python into a more efficient compiled language such as Rust. In this post I explore nucleotide k-mer counting as an example algorithm and evaluate the performance improvement of a few different implementations. Ultimately, my fastest Rust+Python implementation is only 2.5x faster than a pure Python equivalent.
Table of Contents
1. Background and Motivation
In our lab, we often process hundreds of thousands, if not millions, of sequencing reads generated from a malignant tumor sample. These reads are footprints of the mutational landscape underlying the tumor and clinically actionable insights are grounded in understanding these footprints. To view the landscape, we usually piece together sequencing reads to solve a puzzle - a large puzzle in fact. The original picture we are after is typically composed of around 3 billions letters and we have millions of pieces to solve. This is where bioinformatics softwares come to our aid.
Many bread-and-butter bioinformatics softwares are written in “low-level” languages such as C and C++ to deliver fast computation times. These languages provide instructions more native to a computer machine’s hardwares compared to “high-level” languages such as Python. While low-level languages guarantee unparalleled computation times, the development process can prove to be slow and tedious, and the eventual codebase can become quite verbose. This makes the codebase hard to maintain and to deploy over iterations. This is why a versatile and succinct high-level language like Python remains one of the most popularly used programming languages. However, the sheer versatility and succinctness of Python are also sources of bottlenecks of its less-than-ideal execution time. For many clinical settings where speed is crucial to generate timely clinical decisions, Python quickly hits a practical limit. For example, k-mer counting is a common computation in bioinformatics that involves updating a hash table and this simple task written in C++ is 25 times faster than Python.
About a decade ago, a new low-level programming language called Rust was released. In recent years, we have seen an uptick in the number of bioinformatics tools developed in Rust. I have recently started learning Rust and have tried to wrap it in Python so that we can integrate the best of both worlds - the comprehensive suite of statistical and optimization libraries offered in Python and the swift computation time of Rust. This post recounts the story of my recent experience in attempting a number of different technical implementations wrapping Rust in Python and how I landed the one that I like (for now). I also share all the different implementations that I have tried in hopes of saving another bioinformatician’s (or developer’s) time.
To compare different implementations, I chose to work with the k-mer counting problem. For example, given the following 3 sequences “CATGATCCAA”, “AGGATAAGCC”, “GGATCACGAC”, we want to count the occurrence of all 3-mers in these sequences over sliding windows (e.g. "GAT” appears 3 times in the three sequences; “CATGATCCAA”, “AGGATAAGCC”, “GGATCACGAC”).
2. Implementations
Each implementation is composed of three parts:
Generation of tasks (chunks of sequences).
K-mer counting by task workers.
Merging hash tables into one.
For full implementations, please check out here.
2-1. Multiprocessing in Python (Baseline)
To establish the baseline, I first implemented k-mer counting in Python3 as a multi-processing program with the function worker_py defined as the task worker:
count_kmers_python_multiprocess.pyimport multiprocessing import pysam import time import pandas as pd from functools import partial from collections import Counter def worker_py(sequence: str, k: int) -> Counter: """ Fetches k-mers in a given sequence. Parameters ---------- sequence : Sequence. k : k-mer k. shared_list : Shared list. """ counter = Counter() for i in range(0, len(sequence) - k + 1): counter[sequence[i:i+k]] += 1 return counter def count_kmers_python_multiprocess(fasta_file: str, num_processes: int, sequence_chunk: int, k: int, chromosomes: list) -> pd.DataFrame: """ Counts all possible k-mers in a FASTA file. Parameters ---------- fasta_file : FASTA file. num_processes : Number of processes. sequence_chunk : Sequence chunk. k : k-mer k. chromosomes : Chromosomes to count. Returns ------- df_kmers : DataFrame of k-mers with the following columns: 'k_mer', 'count' work_duration : Duration of multiprocessing compute """ # Step 1. Create tasks fasta = pysam.FastaFile(fasta_file) sequences = [] for chrom in chromosomes: chrom_len = fasta.get_reference_length(chrom) for i in range(0, chrom_len, sequence_chunk): curr_start = i curr_end = curr_start + sequence_chunk if curr_end > chrom_len: curr_end = chrom_len curr_chromosome_seq = fasta.fetch( reference=chrom, start=curr_start, end=curr_end ) sequences.append(curr_chromosome_seq.upper()) # Step 2. Run pool = multiprocessing.Pool(num_processes) start_time = time.time() results = pool.map(partial(worker_py, k=k), sequences) end_time = time.time() # Step 3. Merge dictionaries into one outputs = results[0] for curr_result in results[1:]: outputs.update(curr_result) df_kmers = pd.DataFrame.from_dict(outputs.items()) df_kmers.columns = ['k_mer', 'count'] df_kmers.sort_values(['count'], ascending=False, inplace=True) return df_kmers, end_time - start_time
Terminal output with the following parameters:
hg38.fa6 processes1,000,000 nucleotide chunks9-mers (k = 9)chr21
Python implementation of k-mer counting k_mer count NNNNNNNNN 6620936 TTTTTTTTT 44007 AAAAAAAAA 41832 TGTGTGTGT 10454 ACACACACA 10170 Program took 14.662775 seconds in total
2-2. Rust Wrapped in Python
To wrap Rust in Python, I used the maturin package in Python. To start writing a Rust function, first initialize by executing the following in the terminal:
maturin init
This generates a lib.rs file in src/ folder and a Cargo.toml file. We will be writing the Rust codes in the lib.rs file and specify any dependencies in the Cargo.toml file. To compile Rust code using maturin, run:
maturin develop
Let’s add compiler optimization to the Cargo.toml file:
[profile.dev] opt-level = 3 [profile.release] debug = 0
For more on maturin, refer to the official maturin user guide.
2-2-1. Python with Multi-threaded Rust (STL HashMap)
For starters, to take advantage of Rust’s native multi-threading capabilities, I first implemented a multi-threaded k-mer counting function:
lib.rsuse pyo3::prelude::*; use rayon::prelude::*; use std::collections::HashMap; #[pyfunction] fn count_kmers_multithread_stl_hashmap(sequences: Vec<String>, k: usize, num_threads: usize) -> Py<PyAny> { let pool = rayon::ThreadPoolBuilder::new() .num_threads(num_threads) .build() .unwrap(); let (tx, rx) = std::sync::mpsc::channel(); for seq in &sequences { let tx = tx.clone(); let seq = seq.to_owned(); pool.spawn(move || { let mut hm : HashMap<String, i32> = HashMap::new(); let end = seq.chars().count() - k + 1; for i in 0..end { *hm.entry(seq[i..i+k].to_owned()).or_insert(0) += 1; } tx.send(hm).unwrap(); }); } drop(tx); // close all senders let results: Vec<HashMap<String, i32>> = rx.into_iter().collect(); // Merge HashMaps let mut hm : HashMap<&str, i32> = HashMap::new(); for curr_hm in &results { for (key, value) in curr_hm.into_iter() { *hm.entry(&key).or_insert(0) += value; } } return Python::with_gil(|py| { hm.to_object(py) }); } #[pymodule] fn scripts(_py: Python, m: &PyModule) -> PyResult<()> { m.add_function(wrap_pyfunction!(count_kmers_multithread_stl_hashmap, m)?)?; Ok(()) }
The above code makes the Rust function count_kmers_multithread_stl_hashmap available as part of a package (named script in my code).
Make sure to append the following line to Cargo.toml file under [dependencies]:
rayon = "1.5.3"
count_kmers_rust_multithread_stl_hashmap.pyimport pysam import time import pandas as pd import scripts def count_kmers_rust_multithread_stl_hashmap(fasta_file: str, num_processes: int, sequence_chunk: int, k: int, chromosomes: list) -> pd.DataFrame: """ Counts all possible k-mers in a FASTA file. Parameters ---------- fasta_file : FASTA file. num_processes : Number of processes to multiprocess FASTA file. sequence_chunk : Sequence chunk. k : k-mer k. chromosomes : Chromosomes to count. Returns ------- df_kmers : DataFrame of k-mers with the following columns: 'k_mer', 'count' work_duration : Duration of multiprocessing compute """ # Step 1. Create tasks fasta = pysam.FastaFile(fasta_file) sequences = [] for curr_chromosome in chromosomes: curr_chromosome_len = fasta.get_reference_length(curr_chromosome) for i in range(0, curr_chromosome_len, sequence_chunk): curr_start = i curr_end = curr_start + sequence_chunk if curr_end > curr_chromosome_len: curr_end = curr_chromosome_len curr_chromosome_seq = fasta.fetch(reference=curr_chromosome, start=curr_start, end=curr_end) sequences.append(curr_chromosome_seq.upper()) # Step 2. Run start_time = time.time() results = scripts.count_kmers_multithread_stl_hashmap(sequences, k, num_processes) end_time = time.time() # Step 3. Merge dictionaries into one df_kmers = pd.DataFrame(results.items()) df_kmers.columns = ['k_mer', 'count'] df_kmers.sort_values(['count'], ascending=False, inplace=True) return df_kmers, end_time - start_time
2-2-2. Python with Multi-threaded Rust (FX HashMap)
The above multi-threaded Rust implementation resulted in about 55% reduction (i.e. 2.2x improvement) in execution time from the Python implementation. As I looked to further improve the speed of the Rust code, I stumbled upon this page that explains how the HashMap included in the standard library (STL) of Rust implements cryptographically safe hashing. The natural move was to replace the STL HashMap with another HashMap that relies on a faster hashing function, such as rustc-hash (henceforth referred to as FX HashMap). So all of the HashMap declarations in the above Rust code was replaced with the following:
let mut hm : FxHashMap<String, i32> = FxHashMap::default(); let results: Vec<FxHashMap<String, i32>> = rx.into_iter().collect(); let mut hm : FxHashMap<&str, i32> = FxHashMap::default();
Make sure to append the following line to Cargo.toml file under [dependencies]:
rustc-hash = "1.1.0"
Replacing the HashMap implementation from the STL function to the FX version indeed resulted in a noticeable improvement in speed (data not shown).
2-2-3. Python with Multi-processed Rust (STL or FX HashMap)
Then it occurred to me that using pointers in Rust might lead to another boost in speed. After tinkering around for some time, I realized that implementing a pointer-based access to a HashMap in a multi-threaded Rust program is not so straight forward and elegant. Instead, I opted to multiprocessing a Rust function but allowing pointer-based access and update to the HashMap. Shown below is the Rust code for the task worker function:
#[pyfunction] fn count_kmers_fx_hashmap_pointer(sequence: String, k: usize) -> Py<PyAny> { let mut hm : FxHashMap<&str, i32> = FxHashMap::default(); let end = sequence.chars().count() - k + 1; for j in 0..end { *hm.entry(&sequence[j..j+k]).or_insert(0) += 1; } return Python::with_gil(|py| { hm.to_object(py) }); }
Here is a simple update to the Python implementation to include the rust worker function:
results = pool.map(partial(scripts.count_kmers_fx_hashmap_pointer, k=k), sequences)
3. Benchmark Test Results
To systematically benchmark the various implementations, I set up a test to count all 9-mers in chromsome 1 of hg38. I ran the above implementations along with some other variations for comparison purposes. In short, of the 7 implementations, the multi-threaded Rust with FX HashMap resulted in the fastest compute time of 16.828 seconds on average. In contrast, Python took 42.321 seconds on average (Figure 1).
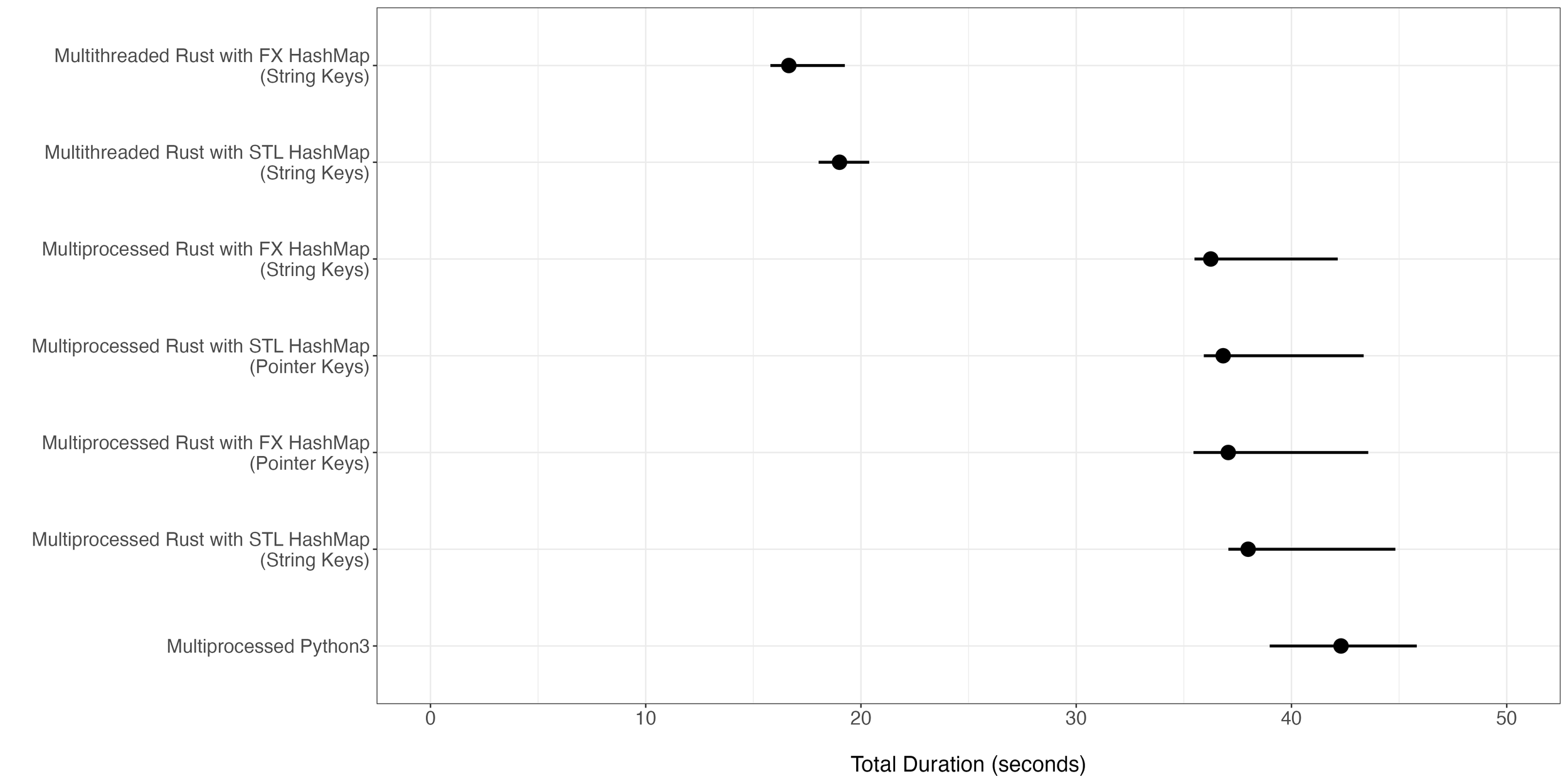
Figure 1. Total execution time of different implementations for the task of 9-mer counting for hg38 chromosome 1. For each implementation, 20 iterations were run with each iteration using 24 cores. The dot in the middle of each bar represents the median total duration while the ends of the bar represent the minimum and the maximum duration time observed.
It should be noted that the two multi-threaded Rust implementations depend on Rust to merge the HashMaps into one. Computation of task workers (step 2) is a subset of all computations accounted for in the total duration. When we just capture the duration of task workers in multi-threaded and multi-processed implementations, we see there are relatively smaller improvements in speed (Figure 2). Based on the duration of task workers in either multi-threaded or multi-processed implementations, we see that HashMap merging (step 3) accounts for an overwhelming fraction of the time saved (Figure 2). In other words, the multi-threaded implementation comes ahead of the multi-processed implementation because of the heavy-lifting performed by Rust in merging HashMaps.

Figure 2. Average execution times of each of the three steps observed in the same tests for Figure 1.
4. Conclusions
In this post, we saw how to wrap Rust in Python. In conclusion, multi-threaded Rust (with FX HashMap) wrapped in Python yielded the fastest implementation with approximately 2.5x improvement in speed over the baseline Python implementation. In the tech world, people often talk about delivering a 10x improvement for a new technology to tip the scale and be widely adopted. Although the speed improvement is clear, I am not convinced that the additional overhead of keeping Rust codebase along with the Python codebase justifies for mere 2.5x improvement in speed. Stay tuned for more on this topic!
Special thanks to Dhuvi Karthikeyan for his invaluable feedback on my post and for suggesting the title of this blog post.